







Syndrome de Usher : quels espoirs de traitements ?

Suite de notre interview dédiée au syndrome de Usher auprès du Pr Isabelle Audo.
Relire la première partie de l'interview : "Syndrome de Usher, une urgence thérapeutique">

Pr Isabelle Audo,
médecin chercheuse spécialisée dans les maladies rétiniennes génétiques,
professeure en ophtalmologie à Sorbonne Université,
directrice adjointe de l’Institut de la Vision,
coordinatrice du Centre de référence Maladies Rares à l’Hôpital National des 15-20. ©DR
Quelles actions pour freiner le syndrome de Usher ?
Il existe cependant un certain nombre de recommandations qui permettent de ralentir l’évolution de la pathologie. « Nous insistons beaucoup sur ces aspects, en s’appuyant sur la logique, mais aussi sur ce qu'on connait d'autres maladies. On conseille ainsi d'éviter ce qui est « toxique » » souligne le Pr Isabelle Audo. Il s’agit notamment du port de verres protecteurs avec des filtres marron-orangé, d’une casquette ou d’un chapeau à visière en extérieur, pour se protéger des lumières de longueur d'onde verte et bleue. Il est également recommandé d’arrêter le tabac : « c’est mon grand combat. On l’oublie toujours parce que c'est le B.A.B.A. Mais il y a plein de patients qui viennent nous voir et qui disent « moi je veux savoir ce qu'il y a à faire en termes de recherche thérapeutique ». Je leur réponds « la première chose à faire, c'est d'arrêter de fumer parce que c'est probablement ce que vous ferez de plus efficace » martèle la médecin.
Autre levier d’action : la nourriture. Pour une personne atteinte du syndrome de Usher, il est en effet essentiel d’enrichir son alimentation en antioxydants. On pensera notamment à tous les aliments riches en lutéine, qu’on retrouve surtout dans les légumes verts. En effet, la lutéine, et la zéaxanthine, sont des pigments naturels situés au centre de la macula et qui sont protecteurs. « Le chou frisé cru est celui qui contient le plus de lutéine. Mais ce n'est pas forcément ce qui est le plus appétissant, il faut le préparer… » sourit Isabelle Audo. Les patients vont être encouragés à manger beaucoup de légumes verts, de fruits et de poissons gras, riches en oméga 3, en évitant les fritures, pour avoir le maximum d'antioxydants par l'alimentation.

Microscopie électronique à balayage d'un œil d'une larve de xénope, illustrant les différentes couches de la rétine
dont la couche des photorécepteurs, colorée en violet. © C. Schietroma, Institut Pasteur. DR
Quels espoirs pour la thérapie génique ?
Quel que soit le gène muté identifié, lors du déclenchement de la rétinopathie pigmentaire du syndrome de Usher, on constate d’abord une dégénérescence des bâtonnets puis des cônes. S’il reste encore assez de photorécepteurs, une approche par thérapie génique peut-être envisagée. Elle fait cependant face à d’importants obstacles, au premier rang desquels, la taille des gènes impliqués. En effet, les deux gènes principaux impliqués dans le syndrome de Usher, MYO7A et USH2A, sont de très grands gènes. Or, actuellement, la thérapie génique nécessite un vecteur viral, qui va rentrer dans le noyau des cellules rétiniennes et y libérer la séquence du gène normal, qui pourra ensuite s’intégrer au matériel génétique de la cellule. Les virus classiquement utilisés en thérapie génique sont ce qu'on appelle les AAV, les adeno-associated virus. Aussi performants soient-ils, ils ont une capacité d'empaquetage limitée, et ne sont pas en mesure d’emballer MYO7A, et encore moins USH2A, qui est encore beaucoup plus grand.
Cependant, des sociétés de biotechnologies travaillent en ce moment même sur des approches alternatives, utilisant deux vecteurs viraux, chacun portant la moitié de la séquence du gène à remplacer, et s’appuyant sur un système de biologie moléculaire qui permettrait la reconstruction du gène entier. « On parle de thérapie par remplacement de gène. Ce serait l’idéal car cette approche cible vraiment la cause de la maladie : le gène est muté, la protéine résultante ne fonctionne plus, alors on ramène la séquence normale pour restaurer la fonction » explique le Pr Isabelle Audo.
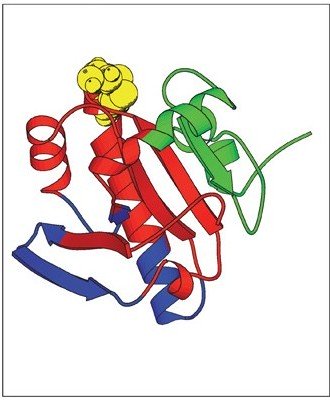
Modèle structurel du RdCVF, facteur de survie des cônes actuellement impliqué dans l'étude clinique Prodigy. © DR
D'autres pistes thérapeutiques en exploration
Un autre espoir pourrait se révéler du côté de la neuroprotection. Le Pr José-Alain Sahel et le Dr Thierry Léveillard ont en effet montré que, dans de nombreuses formes de rétinite pigmentaire (mais pas celles associées à MYO7A ni USH2A), les mutations génétiques sont uniquement exprimées dans les batônnets, et pourtant les cônes disparaissent également. Leur idée a donc été que les batônnets sont essentiels pour apporter un support de survie aux cônes, et que s’ils disparaissent, les cônes dégénèrent secondairement. Leurs recherches ont permis l’identification d’un certain nombre de facteurs, dont un, le Rod derived Cone Viability Factor (RdCVF) pour lequel une étude clinique, appelée Prodigy, est en cours. Si elle n’inclut pas, pour l’instant, de patients Usher, cette étude pourrait mener à la validation d’un mécanisme de neuroprotection universel pour maintenir, voire même restaurer la vision.
Cependant, cette approche implique là encore qu’il reste suffisamment de photorécepteurs pour restaurer leur défaut génétique, donc une prise en charge suffisamment tôt dans la progression de la maladie. Lorsque la majorité des photorécepteurs a disparu, les voies qui restent à explorer sont celles de la restauration visuelle, notamment de l’optogénétique, qui cherche à rendre sensibles à la lumière les cellules situées en aval des photorécepteurs dans la rétine, et de la médecine régénérative. Cette dernière, qui s’appuie sur la greffe de cellules rétiniennes, est cependant encore un lointain espoir. Grâce aux travaux menés dans différentes équipes de recherche, la greffe de cellules de l’épithélium rétinien pigmentaire est maintenant une réalité. Mais le syndrome de Usher nécessite la greffe et l’intégration de photorécepteurs, une opération encore plus complexe, et pour laquelle il reste encore de nombreux points à résoudre. L’équipe du Dr Olivier Goureau, à l’Institut de la Vision, travaille sur ce type de projets depuis des années. « Je dis toujours à mes patients « chaque année je demande à Olivier quand est-ce qu’on greffe des cônes, et chaque année il me répond : « dans dix ans ». Même si bien sur ce sont des choses qui peuvent évoluer très vite s’il y a une modification technologique » commente le Pr Isabelle Audo. Celle-ci souligne l’urgence thérapeutique à être plus innovant et à apporter plus que de la réhabilitation visuelle à ces patients atteints de Usher de type 1, qui se sont ajustés sur le plan auditif en développant la langue des signes ou en bénéficiant des implants cochléaires, mais qui sont à risque de perdre la vue.
Propos recueillis par Aline Aurias.
Soutenez la recherche , soutenez l'Institut de la Vision >

À lire aussi

Syndrome de Usher, une urgence thérapeutique
Forme la plus courante de surdicécité, le syndrome de Usher fait partie de ces maladies génétiques rares pour lesquelles la science n’a pas encore de traitement.

Les troubles de la vision et les maladies des yeux de A à Z

Le prix Wolf de médecine attribué au Pr José-Alain Sahel et au Dr Botond Roska
Le Pr José-Alain Sahel a reçu le prestigieux prix Wolf de médecine avec son collègue Dr Botond Roska, docteur en médecine et es sciences, de l'Institut d'ophtalmologie moléculaire et clinique de Bâle (IOB), pour leurs travaux visant à restaurer la vue à l'aide de l'optogénétique, un outil de rech

13 avancées majeures à l'Institut de la Vision
Si le temps de la recherche paraît souvent très long, et tout particulièrement pour les patients atteints de pathologies ne disposant d'aucun traitement, la recherche sur les maladies de la vision a